Note
Go to the end to download the full example code
6.4. Converter benchmarking¶
Converter have a default method.
Notem however, that several methods may be available. Moreover, you may have a method that you want to compare with the implemented one. To do so you will need to implement your method first. Then, simply use our benchmarking framework as follows.
from bioconvert import Benchmark
from bioconvert import bioconvert_data
from bioconvert.bam2cov import BAM2COV
from bioconvert.fastq2fasta import FASTQ2FASTA
Get the convert you wish to benchmark
input_file = bioconvert_data("test_measles.sorted.bam")
conv = BAM2COV(input_file, "test.cov")
# input_file = bioconvert_data("test_fastq2fasta_v1.fastq")
# conv = FASTQ2FASTA(input_file, "test.fasta")
Get the Benchmark instance
bench = Benchmark(conv)
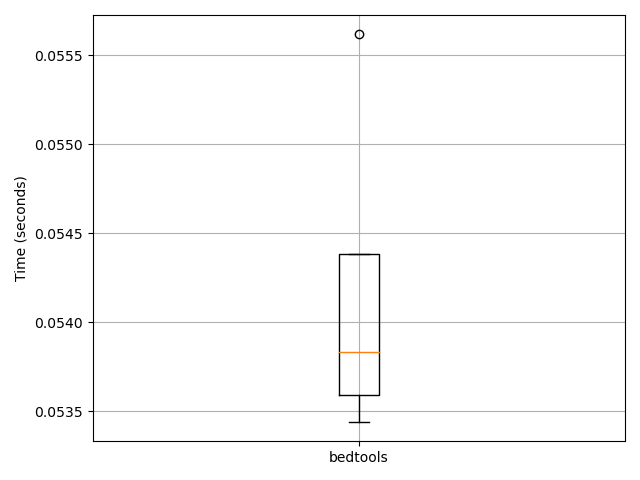
bench.plot()
# You can now see the different methods implemented in this
# converter and which one is the fastest.
Evaluating method bedtools: 0%| | 0/5 [00:00<?, ?it/s]
Evaluating method bedtools: 40%|#### | 2/5 [00:00<00:00, 18.17it/s]
Evaluating method bedtools: 80%|######## | 4/5 [00:00<00:00, 18.38it/s]
Evaluating method bedtools: 100%|##########| 5/5 [00:00<00:00, 18.37it/s]
{'time': {'bedtools': [0.055616140365600586, 0.05438423156738281, 0.05359005928039551, 0.053835391998291016, 0.05344033241271973]}, 'CPU': {'bedtools': [37.5, 41.7, 40.0, 36.4, 40.0]}, 'memory': {'bedtools': [15.9, 15.9, 15.8, 15.8, 15.8]}}
Total running time of the script: ( 0 minutes 0.390 seconds)